

PCV2 entwickelte sich zu einer der verheerendsten viralen Infektionen in Schweinebetrieben und hatte aufgrund der direkten Schäden und Kosten der Bekämpfung erhebliche wirtschaftliche Auswirkungen. PCV2 ist der Krankheitserreger des Postweaning Multisystemic Wasting Syndrome (PMWS - seuchenhaftes Kümmern nach dem Absetzen), das heutzutage als PCV2-bedingte systemische Erkrankung (PCV2-SD, Abb. 1) bezeichnet wird. Epidemiologische und experimentelle Studien haben bestätigt, dass die genetische Vielfalt, die gekennzeichnet ist durch weltweit auftretende Wellen neuer Genotypen und die Zirkulation rekombinanter Stämme, möglicherweise die Virulenz von PVC2 beeinflusst. Möglicherweise ist dies der Grund dafür, dass die intraspezifische Klassifizierung von PCV2 früher immer kontrovers diskutiert wurde. 2008 schlug das EU-Konsortium für PCV2-Erkrankungen eine standardisierte Nomenklatur für die Definition der PCV2-Genotypen auf Grundlage paarweiser Sequenzvergleiche vor (www.pcvd.eu). Die Analysen, die bezüglich der vollständigen und Kapsid-Nukleotidsequenzen (ORF2) von PCV2 durchgeführt wurden, bestimmten zwei Distanz-Schwellenwerte bei 0,020 bzw. 0,035. Wenn die p-Distanz zwischen zwei Sequenzen größer war als diese Werte, ging man davon aus, dass die Stämme zu zwei verschiedenen Genotypen gehörten. Als Ergebnis dieser Analysen wurden die vier Genotypen PCV2a, PCV2b, PCV2c und PCV2d (auch als PCV2b-Mutante bekannt) identifiziert.


Abbildung 1: Drei Monate altes Schwein mit PCV2-bedingter systemischer Erkrankung: Auffällig sind das deutlich zu sehende Rückgrat als Hinweis auf die Wachstumsretardierung und die Blässe (Anämie).
Seit 2008 wurde eine große Anzahl an PCV2-Sequenzen in die GenBank (www.ncbi.nlm.nih.gov) aufgenommen und es wurden verschiedene neue Genotypen vorgeschlagen, obwohl diese nicht immer bestätigt werden konnten. Ein erheblicher Teil dieser Sequenzen ist rekombinanten Ursprungs und in manchen Fällen zirkulierten sie mit zunehmender Prävalenz in verschiedenen asiatischen Ländern und in den USA. In einer Abhandlung, die vor kurzem veröffentlicht wurde (Franzo et al, 2015), analysierte ein internationales Team von Wissenschaftlern erneut die intraspezifische Taxonomie von PCV2 und die Definition der Genotypen, um deren aktuelle Gültigkeit zu überprüfen und die Nomenklatur zu vereinheitlichen und letztendlich weitere Irrtümer zu vermeiden.
Die erhaltenen Ergebnisse deuten darauf hin, dass bei den Kapsid- und Genom-Sequenzen von PCV2, die derzeit zur Verfügung stehen, einige Voraussetzungen der Methode missachtet wurden, die vor fast zehn Jahren der Definition der Schwellenwerte diente. Insbesondere hat sich die Annahme gleicher Nukleotid-Substitutionsraten der verschiedenen PCV2-Genotypen nicht bewahrheitet. Außerdem erschien die Definition eines einzigen Cut-off-Werts zur Definition der PCV2-Genotypen kompliziert. Zudem wären die Schwellenwerte, die zur Definition der Genotypen vorgeschlagen wurden, ohne Zweifel sinnlos (Abb. 2). Diese Hinweise haben zusammen mit den Unmengen neuer bekannter Sequenzen beträchtliche Auswirkungen auf die intraspezifische Klassifizierung von PCV2. Demzufolge sind die Schwellenwerte, die seit 2008 zur Definition der PCV2-Genotypen gelten, derzeit nicht für alle PCV2-Stämme anwendbar, weshalb diese Methode neu überdacht werden sollte.
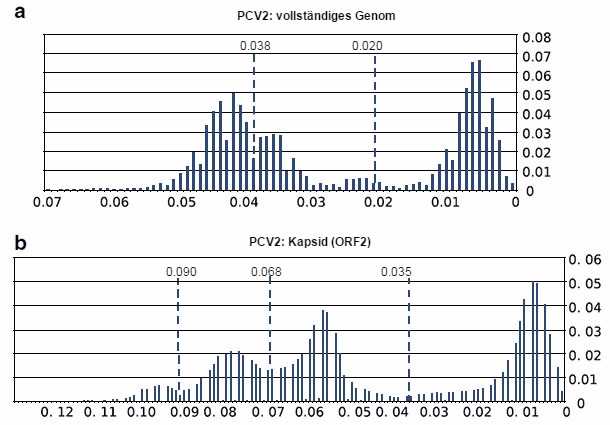
Abbildung 2: Analysen der Schwellenwerte vollständiger PCV2-Genome (a) und von ORF2-Sequenzen (b). Festgestellt wurde der Anteil der paarweisen p-Distanzen innerhalb eines p-Distanz-Intervalls von 0,01. Die vorgeschlagenen Schwellenwerte für das vollständige Genom (0,020), das Kapsid (0,035) sowie die sich aus der erneuten Analyse ergebenden Schwellenwerte (0,038, 0,068 und 0,090) werden ebenso angegeben.
Demgemäß wird eine alternative Methode zur Genotypisierung der PCV2-Stämme eindeutig empfohlen. Der vorgeschlagene Ansatz berücksichtigt das Vorkommen unterschiedlicher rekombinanter Stämme, die tatsächlich zu mehr als einem Genotyp gehören. Angesichts der hohen Rekombinationshäufigkeit, von der berichtet wurde, zog man einen Klassifizierungsansatz auf Grundlage des ORF2-Gens vor. Mit dem Ziel, ein eindeutiges Klassifikationsschema anzubieten, wurden mehrere Referenzsequenzen ausgewählt, deren Klassifizierung hinsichtlich der phylogenetischen Methoden eindeutig war. Dies erlaubt uns, mithilfe der phylogenetischen Rekonstruktion und 47 genotypspezifischer Marker-Nukleotidpositionen vier Genotypen zu definieren. Diese Markerpositionen charakterisieren durchweg (>95%) jeden Genotyp und können als Referenz benutzt werden, um eine Problemsequenz einem bestimmten Genotyp zuzuordnen. Die vorgeschlagene Klassifizierungsmethode ist nicht nur robust, sondern auch sehr schnell und einfach durchzuführen.
Die taxonomische Einordung von PCV2 ist dennoch eine echte Herausforderung. Die Ergebnisse, von denen berichtet wurde, bestätigen die Variabilität der viralen Sequenzen und die hohe intra- und intergenotypische Rekombinationshäufigkeit zwischen PCV2 Stämmen, was die Schwierigkeiten aufzeigt, sich auf eine eindeutige Definition des Genotyps für dieses Virus festzulegen. Zudem ist die Wahrscheinlichkeit des Auftretens neuer PCV2-Genotypen in Zukunft sehr hoch und neue, robuste Methoden zur Genotypisierung könnten zu einem dringend benötigten Instrument werden.
Die Klassifizierung von PCV2 in Genotypen ist aus praktischen Gründen relevant. Bislang beruhten alle auf dem europäischen und nordamerikanischen Markt verfügbaren Impfstoffe auf einem PCV2a-Genotyp, obwohl die Genotypen PCV2b und PCV2d am weitesten verbreitet sind. Obwohl sich ein hohes Maß an Kreuzimmunität unter diesen drei Genotypen gezeigt hat, wäre es sehr interessant festzustellen, ob die Wirksamkeit des Impfstoffs gegenüber all diesen unterschiedlichen Genotypen gleich ist. Außerdem deuten einige phylogenetische Studien, die bisher veröffentlicht wurden, darauf hin, dass bestimmte Änderungen der Aminosäuren bei PCV2-Sequenzen, die aus geimpften und ungeimpften Betrieben stammen, auftreten können. Tatsächlich wurde über die Möglichkeit spekuliert, dass der potentielle Ausbruch mutanter Viren in Zukunft aufgrund dieses Impfdrucks hervorgerufen werden könnte. Eine sachgerechte Überwachung der Genotypen ist deshalb entscheidend für die Beurteilung der Wirksamkeit des Impfstoffs.

