

Das Porcine Reproduktive und Respiratorische Syndrom (PRRS) ist nach wie vor die Krankheit mit den bei weitem stärksten wirtschaftlichen Auswirkungen in der Schweineindustrie und seine Bekämpfung ist alles andere als zufriedenstellend. Ein besseres und umfassenderes Bild der PRRSV-Variation und der Überwachung der Zirkulation neuer Stämme in einem bestimmten Gebiet/ Land/ Kontinent würde Tierärzten und Erzeugern sicherlich dabei helfen, Kontroll- und möglicherweise auch Tilgungsprogramme durchzuführen. Zu diesem Zweck stand ab den späten 1990er Jahren die PRRSV-Sequenzierung weltweit, aber hauptsächlich in Nordamerika, Europa und Südostasien, zur Verfügung.
Das PRRSV-Genom (s. Bild 1) ist ein einzelsträngiges RNA-Molekül, wodurch es während der Replikation im Wirt anfällig für „Fehler“ (genetische Mutationen) ist. Diese „Tendenz, Fehler zu machen“ führt dazu, dass es im Feld verschiedene PRRSV-Stämme gibt, die bezüglich ihrer eigenen genetischen Sequenz einzigartig sind. Ob diese Sequenzunterschiede zu „unterschiedlichem (klinisch-pathologischem oder immunologischem) Verhalten“ führen, ist unter Fachleuten aus der Praxis und Forschern nach wie vor umstritten.

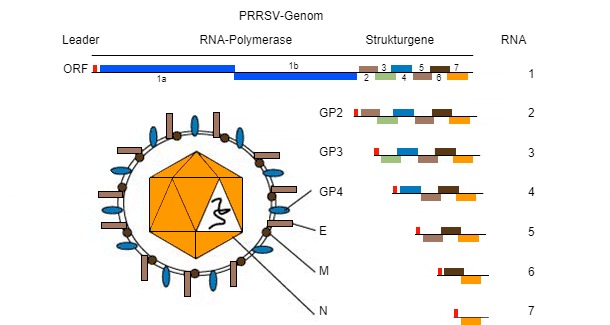
Abbildung 1: Das PRRSV-Genom ist ein einzelsträngiges RNA-Molekül.
Grundlagen der PRRS-Sequenzierung
Die Virus-Sequenzierung erfolgt auf Grundlage von PCR-Produkten von Feldproben (Seren, Gewebe, orale Flüssigkeiten), wobei die Nukleotide normalerweise aus einigen viralen RNA-Genomfragmenten (s. Abbildung 2) in den Zielregionen – ORFs (offener Leserahmen) gelesen werden und dann die prozentuale Homologie durch phylogenetische Analysen mit Hilfe von spezieller Software verglichen wird. Das Ergebnis dieses Prozesses gibt den Ähnlichkeitsgrad (Homologie) zwischen verschiedenen PRRSV-Stämmen wieder. Mit Hilfe von Software zur grafischen Visualisierung kann man auch ein Dendrogramm (oder „phylogenetischen Baum“) erstellen, das die Verwandtschaft (oder Nicht-Verwandtschaft) mit der Sequenz des Referenzvirus zeigt (s. Bild 3).

Abbildung 2: Die Virus-Sequenzierung erfolgt auf Grundlage von PCR-Produkten, wobei die Nukleotide normalerweise aus einigen viralen RNA-Genomfragmenten in den Zielregionen – ORFs gelesen werden.
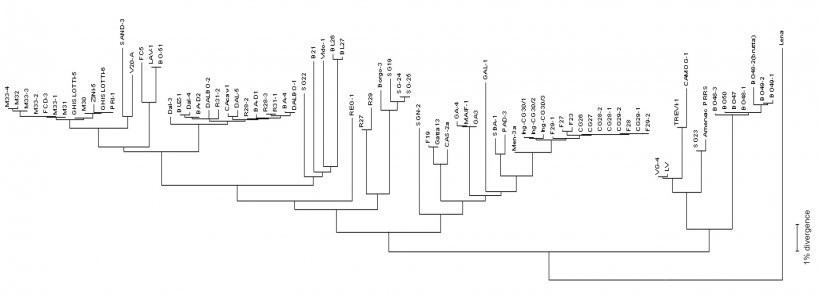
Abbildung 3: Dendrogramme oder „phylogenetische Bäume“ werden benutzt, um den Ähnlichkeitsgrad (Homologie) zwischen verschiedenen PRRSV-Stämmen und einer Referenzvirensequenz grafisch darzustellen.
Das PRRSV-Genom kodiert mindestens 10 ORFs. Die für die Sequenzierung am häufigsten verwendeten ORFs sind ORF5 (Kodierung des nicht glykosylierten E-Proteins) und ORF7 (Kodierung des Nukleokapsid-Proteins (N-Protein)), auch wenn sie nur 4 % bzw. 3 % des gesamten Genoms darstellen. ORF5 stellt eine variablere Region dar, während ORF7 einen stärker konservierten Bereich darstellt. Aus diesem Grund ist ein Variationsgrad (d. h. eine Variation von 5%), der im ORF7 gefunden wird, im Vergleich zum gleichen Variationsgrad im ORF5 hinsichtlich der genetischen Veränderung „dramatischer“. Die Interpretation von Ähnlichkeiten (d. h., ob Viren miteinander verwandt sind oder nicht) erfordert viel mehr zusätzliche Informationen, da die Rate der genetischen Veränderung sehr variabel sein kann.
Es ist äußerst wichtig, eine Protokolldatei aller Sequenzen anzulegen, die eindeutig identifiziert und mit sorgfältigen Anmerkungen zum entsprechenden Datum, Betriebstyp (Betrieb 1-2-3), zur Umstallung der Schweine, zum Standort (GPS-Koordinaten mit Breiten- und Längengrad) und Herkunft der Sequenz (Art des Tieres/ Gewebe/ Probe) versehen sind. Bislang umfasst unsere PRRSV-Sequenzdatenbank mehr als 1.300 ORF7-Sequenzen aus dem Jahr 2002. Um Unterschiede interpretieren und verstehen zu können, ist es umso wichtiger, einzelne Sequenzen mit klinischen Ereignissen wie der Anzahl der Sauen mit Aborten und der Sterblichkeitsrate bis zum Absetzen in den Betrieben 1 und der Sterblichkeitsrate in den Betrieben 2 und 3 zu vergleichen.
Praktische Fragen
Häufig gestellte Fragen von Erzeugern und Tierärzten sind:
- Stellen die beobachteten genetischen Unterschiede zwischen den Sequenzen eine normale Variation eines einzelnen PRRSV-Stamms in einem Betrieb/ System dar oder repräsentieren sie mehrere unterschiedliche Stämme, die in einem Betrieb/ System gleichzeitig oder innerhalb kurzer Zeiträume auftreten?
- Habe ich es gerade mit einem „neuen Ausbruch“ zu tun, der durch einen „neuen Stamm“ verursacht wurde, oder handelt es sich um eine Rezirkulation?
Um diese Fragen zu beantworten, müssen wir uns auf den anerkannten Grad der Übereinstimmung zweier viraler Stämme einigen, die innerhalb einer bestimmten Zeitspanne (12-24 Monate?) erfasst wurden, mit anderen Worten den Cutoff der Ähnlichkeit. Eine Sequenzhomologie von 97-98 % oder ein Unterschied von 2-3 % ist ein allgemein akzeptierter Wert. Nach meiner Erfahrung ist es ziemlich schwierig, in einer „klinisch stabilen geschlossenen Population“ (einem herkömmlichen Sauenbetrieb oder einem Betrieb mit Rein-Raus-Verfahren) eine Veränderung zu erkennen, die über 2 % liegt, da wir beobachteten, dass man den „gleichen Stamm“ in einem einzigen klinisch stabilen Betrieb für einen Zeitraum von 3 Jahren erhält. Im Gegensatz dazu wird jedes Mal, wenn wir eine konstante PRRS-Aktivität feststellten, ein „neuer“ und phylogenetisch andersartiger Stamm (90 % Homologie oder weniger) gefunden. Leider sind wir uns nicht absolut sicher, ob diese großen Unterschiede, die wir manchmal beobachten, das Ergebnis einer plötzlichen Veränderung/ Mutation des Virus (meiner Meinung nach unwahrscheinlich) oder der Einführung eines neuen Stamms sind. Was eindeutig und allgemein anerkannt ist, ist die Tatsache, dass die genetische Ähnlichkeit/ Vielfalt keinerlei Voraussagen für die immunologische Ähnlichkeit (also einen Hinweis auf Kreuzimmunität) oder die intrinsische Pathogenität zulässt (keine Aussage darüber, ob ein bestimmter Stamm „gut“ oder „schlecht“ ist).
Die kompletten Genomsequenzen, die derzeit zur Verfügung stehen (leider mehr für Forschungszwecke als für den täglichen diagnostischen Gebrauch), werden sicherlich dazu beitragen, diese Frage zu beantworten.
Es ist sehr wichtig, neue PRRSV-Sequenzen im Vergleich zu umfangreichen Referenzdaten, die den Betrieb, das System und die Region repräsentieren, sowie die Sequenzen der zur Verfügung stehenden kommerziellen Impfstoffe zu analysieren (dies wird es ermöglichen, zwischen Feld- und Impfstämmen zu unterscheiden). Im Moment benutzen wir immer noch viel Open-Source-Software, die von der Universität Padova verwaltet wird, um unsere phylogenetischen Bäume zu erstellen, die entsprechend dem Durchlauf der Schweine innerhalb unseres gesamten Produktionssystems organisiert sind. Wir könnten auch bald zwei weitere „Ad-hoc-Computerprogramme“ (Bioportal von der Universität von Kalifornien, Davis und CLASSIFARM-Path von IZSLER (Brescia, Italien) nutzen, die eine viel größere Menge an Sequenzen zum Vergleichen haben, was dazu beitragen würde, die Zirkulation von PRRSV in Italien und möglicherweise in der EU besser zu verstehen.
Danksagungen: Mein Dank gilt Prof. Michele Drigo (UNI-PD) für die interessante Diskussion und Revision dieser Arbeit.




